NCLEX pathophysiology is the conceptual infrastructure that makes clinical reasoning possible. Without understanding why a patient with left-sided heart failure develops pulmonary crackles, a candidate can memorize that crackles are a left heart failure finding — but cannot reason through a novel question where the clinical presentation is partially obscured, where two options both mention respiratory findings, or where the question asks what the nurse anticipates next as the condition progresses. Pathophysiology converts isolated clinical facts into mechanistic understanding that applies to any presentation of a condition, familiar or novel.
The challenge with NCLEX pathophysiology is scope. Every body system involves complex mechanisms, and the NCLEX tests application of those mechanisms rather than recall of them. A candidate who understands that decreased cardiac output in heart failure activates the renin-angiotensin-aldosterone system — which causes sodium and water retention, which worsens volume overload, which further stresses the failing heart — can answer questions about diuretics, dietary sodium restriction, weight monitoring, and fluid management that never explicitly mention pathophysiology at all. The mechanism explains the nursing actions, making them logically derivable rather than separately memorizable. This is the clinical reasoning the NCLEX rewards, and it is what NCLEX pathophysiology understanding builds. This guide covers the core NCLEX pathophysiology mechanisms across the six highest-yield body systems: cardiovascular, respiratory, neurological, renal, endocrine, and immune and inflammatory response. For each system the focus is not on encyclopedic detail but on the specific mechanisms that explain the clinical presentations, complications, and nursing interventions most consistently tested on the exam. Understanding these mechanisms at the level this guide presents them is what converts NCLEX pathophysiology from a memorization burden into a clinical reasoning foundation.
Why Pathophysiology Mastery Changes Everything in NCLEX Preparation
Before covering individual system mechanisms, understanding why NCLEX pathophysiology understanding specifically improves exam performance — rather than simply expanding clinical knowledge — clarifies the preparation approach that makes pathophysiology study most efficient.
From Facts to Mechanisms: The Clinical Reasoning Difference
NCLEX pathophysiology knowledge converts clinical facts from isolated, separately memorizable items into a connected network where each element is logically derivable from a small number of foundational mechanisms. A candidate who memorizes that heart failure patients receive furosemide, need daily weights, follow a sodium-restricted diet, and should report a 2 kg weight gain has four disconnected facts. A candidate who understands that heart failure reduces cardiac output, triggering compensatory fluid retention via RAAS activation and ADH release, which produces volume overload that worsens cardiac work and causes pulmonary and peripheral edema — has one mechanism from which all four facts are logically derivable. If they forget that daily weight is the monitoring tool, they can derive it: fluid retention produces weight gain before visible edema appears, making weight the most sensitive early fluid retention indicator. This derivability from mechanism is what allows clinical reasoning to function correctly under novel question presentations where memorized fact patterns do not map cleanly onto the scenario.
Pathophysiology as a Clinical Prediction Engine
One of the most powerful preparation applications of NCLEX pathophysiology is clinical prediction — the ability to anticipate what will happen next in a clinical scenario based on the mechanism of the underlying condition. The NCLEX action verb the nurse anticipates tests this skill directly, as do questions about which complications the nurse monitors for, which assessments are most important as the condition progresses, and which changes in clinical data signal deterioration versus improvement. A candidate who understands that increasing intracranial pressure reduces cerebral perfusion pressure by compressing venous outflow can predict that the early signs will be subtle cognitive changes and headache, that later signs will involve pupillary changes as cranial nerve compression occurs, and that the Cushing triad of hypertension, bradycardia, and irregular respirations represents late herniation in progress — all derivable from the mechanism without memorizing each sign separately as an unconnected list.
Cardiovascular Pathophysiology: Pumps, Pressure, and Perfusion
Cardiovascular NCLEX pathophysiology is the highest-yield system on the exam. Every cardiovascular nursing action — fluid management, medication administration, monitoring priorities — is logically connected to the hemodynamic mechanisms of the underlying condition.
Heart Failure: The Compensatory Spiral
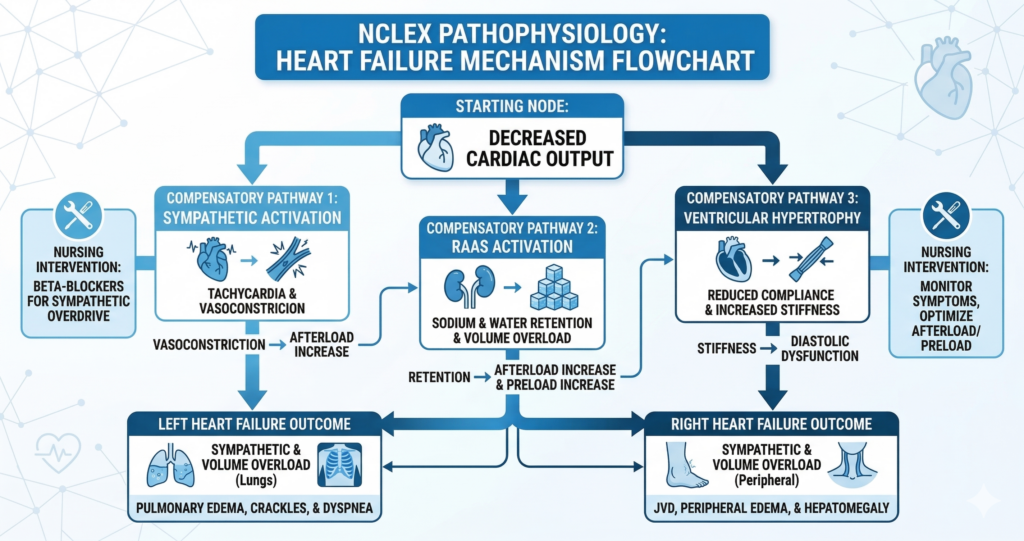
Heart failure NCLEX pathophysiology begins with decreased myocardial contractility or impaired filling, which reduces cardiac output. Reduced cardiac output triggers three compensatory mechanisms that provide short-term benefit but produce long-term harm. First, the sympathetic nervous system activates — increasing heart rate and peripheral vasoconstriction to maintain blood pressure. Second, the renin-angiotensin-aldosterone system activates — causing sodium and water retention to increase preload and support cardiac output. Third, the myocardium undergoes hypertrophy — the heart muscle thickens and enlarges to compensate for increased workload. Each compensatory mechanism produces clinical findings: sympathetic activation causes tachycardia and elevated blood pressure early; RAAS activation produces edema, weight gain, and increased preload; hypertrophy eventually reduces ventricular compliance and worsens filling. Left-sided failure causes fluid back-pressure into the pulmonary circulation, producing pulmonary edema — crackles, dyspnea, orthopnea, pink frothy sputum. Right-sided failure causes fluid back-pressure into the systemic venous circulation, producing jugular venous distension, peripheral edema, and hepatomegaly. Every nursing intervention for heart failure addresses one or more of these mechanisms: diuretics reduce volume overload; ACE inhibitors block RAAS; beta-blockers reduce sympathetic activation; sodium restriction reduces RAAS-driven fluid retention.
Atherosclerosis and Acute Coronary Syndrome
The NCLEX pathophysiology of acute coronary syndrome begins with atherosclerotic plaque formation in coronary arteries — lipid deposition beneath the endothelial lining that progressively narrows the arterial lumen and reduces coronary perfusion. When a plaque ruptures, platelet aggregation and thrombus formation produce sudden severe coronary occlusion. Partial occlusion producing transient ischemia without cell death is unstable angina. Complete occlusion lasting long enough to cause cardiomyocyte death is myocardial infarction. The zone of injured but not yet dead myocardium surrounding the infarct core — the ischemic penumbra — is the target of time-critical revascularization: restoring coronary flow through PCI or thrombolytics salvages myocardium that would otherwise die. Every minute of delay converts ischemic penumbra into necrotic infarct, which is why time-to-treatment is the highest-priority nursing concern in NCLEX pathophysiology ACS questions. The nursing actions of aspirin administration (platelet aggregation inhibition), nitroglycerin (coronary vasodilation), and oxygen only if saturations fall below 94 percent are all directly derivable from this mechanism.
Shock: The Perfusion Failure Framework
Shock NCLEX pathophysiology is unified by one mechanism regardless of type: inadequate tissue perfusion relative to metabolic demand, producing cellular hypoxia and progressive organ dysfunction. Hypovolemic shock results from inadequate circulating volume — from hemorrhage, severe dehydration, or plasma loss. Distributive shock results from vasodilation distributing blood away from vital organs — septic shock from inflammatory mediators, anaphylactic shock from histamine release, neurogenic shock from loss of sympathetic vascular tone. Cardiogenic shock results from pump failure — the heart cannot generate sufficient cardiac output to maintain perfusion despite adequate volume. Obstructive shock results from mechanical impedance to blood flow — tension pneumothorax, cardiac tamponade, or massive PE preventing forward flow. Despite different causes, all shock types produce the same downstream consequence: inadequate oxygen delivery to tissues, causing anaerobic metabolism, lactic acid accumulation, and progressive organ failure. The NCLEX pathophysiology clinical recognition rule is tachycardia as the earliest sign — the compensatory response to any perfusion threat — followed by hypotension as compensation fails.
Respiratory Pathophysiology: Gas Exchange, Obstruction, and Compliance
Respiratory NCLEX pathophysiology centers on the two fundamental functions of the respiratory system — ventilation (moving air in and out) and gas exchange (oxygen uptake and carbon dioxide elimination) — and the mechanisms by which different conditions impair one or both.
Obstructive vs. Restrictive Lung Disease
Understanding the NCLEX pathophysiology distinction between obstructive and restrictive lung disease explains the different clinical presentations, spirometry findings, and nursing priorities for each category. Obstructive lung disease — COPD, asthma, bronchiectasis — impairs exhalation by increasing airway resistance. Air enters relatively normally but cannot exit efficiently, producing air trapping and hyperinflation. The barrel chest of COPD reflects chronic hyperinflation from air trapping. Respiratory failure in obstructive disease produces elevated CO2 (hypercapnia) because air cannot be exhaled, and the compensatory mechanism of breathing fast and shallow makes things worse by reducing tidal volume and increasing dead space. The NCLEX pathophysiology implication of CO2 retention in chronic COPD is the hypoxic drive — the respiratory center adapts to chronically elevated CO2 and relies on relative hypoxemia as the breathing stimulus, making high-flow oxygen a respiratory depression risk. Restrictive lung disease — pulmonary fibrosis, pleural effusion, pneumonia, ARDS — impairs lung expansion, reducing tidal volume and total lung capacity. Gas exchange fails because alveolar surface area is reduced or consolidated. Respiratory failure in restrictive disease produces hypoxemia without initial hypercapnia because the patient breathes faster and deeper to compensate, washing out CO2 effectively until respiratory muscle fatigue sets in.
Ventilation-Perfusion Mismatch
Ventilation-perfusion mismatch is the core NCLEX pathophysiology mechanism behind several high-yield respiratory conditions. Normal gas exchange requires matching of ventilation (air reaching alveoli) and perfusion (blood flowing through pulmonary capillaries) in the same alveolar units. When ventilation occurs without perfusion — as in pulmonary embolism — alveoli receive fresh air but no blood for gas exchange: dead space ventilation that contributes nothing to oxygenation. When perfusion occurs without ventilation — as in pneumonia consolidation, atelectasis, or pulmonary edema — blood passes through lung tissue that contains no functional air, returning to the systemic circulation un-oxygenated: an intrapulmonary shunt that directly lowers arterial oxygen saturation. The NCLEX pathophysiology clinical implication of shunt physiology is that supplemental oxygen does not fully correct hypoxemia from intrapulmonary shunting — the shunted blood never contacts the oxygen-enriched air. This explains why patients with severe pneumonia or ARDS may have persistently low oxygen saturations despite high-flow supplemental oxygen, and why PEEP in mechanical ventilation — which recruits collapsed alveoli and reduces shunt fraction — improves oxygenation more effectively than simply increasing FiO2.
Pulmonary Embolism: Obstruction and Its Cascade
Pulmonary embolism NCLEX pathophysiology begins with thrombus formation — usually from deep vein thrombosis in the lower extremities — that breaks loose and lodges in a pulmonary artery, obstructing blood flow to a segment of lung. This obstruction creates a ventilation-perfusion mismatch (ventilation without perfusion in the affected segment), increases pulmonary arterial pressure as blood is redirected through remaining vessels, and acutely elevates right ventricular afterload. Severe PE causes right ventricular strain and failure — the mechanism behind the hemodynamic instability of massive PE. The clinical presentation reflects these mechanisms: sudden dyspnea from hypoxia, pleuritic chest pain from pleural irritation adjacent to the infarcted lung segment, tachycardia from sympathetic compensation, and hypoxemia from V-Q mismatch. The classic triad of dyspnea, chest pain, and hemoptysis appears in a minority of cases but is highly specific when all three are present. NCLEX pathophysiology nursing priorities are anticoagulation to prevent clot extension and recurrence — not to dissolve the existing clot, which the body’s fibrinolytic system handles — with thrombolytics reserved for massive PE with hemodynamic compromise.
Neurological Pathophysiology: Pressure, Perfusion, and Electrical Disruption

Neurological NCLEX pathophysiology is dominated by three mechanisms: impaired cerebral perfusion from stroke or increased intracranial pressure, altered electrical activity from seizure disorders, and disrupted autonomic regulation from spinal cord injury. Understanding each mechanism explains the clinical presentations and emergency nursing priorities that appear repeatedly across neurological NCLEX questions.
Stroke: Ischemic Cascade and Hemorrhagic Transformation
Ischemic stroke NCLEX pathophysiology begins with arterial occlusion — from thrombus or embolus — that reduces blood flow to a brain territory below the threshold for neuronal function. The core of the ischemic zone undergoes rapid irreversible necrosis — cell death from energy failure within minutes of occlusion. Surrounding this necrotic core is the ischemic penumbra — a zone of functionally impaired but structurally intact neurons that remain viable for hours if reperfusion is achieved. Time-to-treatment urgency in stroke management is directly explained by this mechanism: every minute of continued occlusion converts penumbral neurons into necrotic ones, shrinking the tissue that reperfusion can salvage. The NCLEX pathophysiology tPA safety rule connects directly: tPA dissolves the occlusive clot, but in hemorrhagic stroke — where the vessel has already ruptured — it would catastrophically worsen bleeding. CT scan before tPA is mandatory to exclude hemorrhage. The blood pressure threshold for tPA administration (below 185/110) prevents reperfusion injury — restoration of blood flow to ischemic tissue under very high pressure can cause hemorrhagic transformation of the infarct.
Increased Intracranial Pressure: The Monroe-Kellie Doctrine
Increased intracranial pressure NCLEX pathophysiology is grounded in the Monroe-Kellie doctrine: the skull is a rigid, non-expandable container whose total contents — brain tissue, cerebrospinal fluid, and blood — must remain constant in volume. When any component increases — from tumor, hemorrhage, edema, or hydrocephalus — the others must decrease to compensate, or intracranial pressure rises. Initial compensation occurs through CSF displacement from the cranium into the spinal subarachnoid space and through venous blood displacement. When these compensatory mechanisms are exhausted, even small additional volume increases produce large ICP rises. Rising ICP reduces cerebral perfusion pressure (the difference between mean arterial pressure and ICP), threatening cerebral ischemia. The NCLEX pathophysiology clinical sequence is: early ICP elevation produces subtle cognitive changes and headache — the earliest signs. As ICP rises further, unilateral pupil dilation occurs from herniation compressing cranial nerve III. Late herniation produces the Cushing triad — hypertension with widening pulse pressure, bradycardia, and irregular respirations — the brainstem’s final attempt to maintain perfusion. Every nursing intervention for raised ICP addresses the Monroe-Kellie mechanism: elevating the head of bed promotes venous drainage; avoiding hip flexion and head rotation prevents venous outflow obstruction; controlling pain and agitation reduces cerebral metabolic demand and blood flow.
Seizures: Abnormal Electrical Synchronization
Seizure NCLEX pathophysiology results from abnormal, hypersynchronous electrical discharge in a population of neurons — triggered by disruptions of the normal balance between excitatory and inhibitory neurotransmission. Anything that lowers the seizure threshold — hypoglycemia, hyponatremia, hypocalcemia, hypoxia, fever, sleep deprivation, or withdrawal from sedatives or alcohol — can precipitate a seizure by sufficiently disrupting this balance. During a tonic-clonic seizure, the tonic phase (muscle rigidity and apnea) is followed by the clonic phase (rhythmic muscle jerking), then the postictal phase (confusion, exhaustion, and headache from metabolic recovery). NCLEX pathophysiology nursing priorities during a seizure protect the patient from injury without restraining the movements or forcing anything into the mouth. Position the patient on their side during the clonic and postictal phases to prevent aspiration. Time the seizure duration — status epilepticus (continuous or rapidly recurring seizures lasting more than five minutes without recovery) is a neurological emergency requiring immediate IV benzodiazepine administration and provider notification.
Renal Pathophysiology: Filtration Failure and Its Systemic Consequences
Renal NCLEX pathophysiology is fundamentally about what happens when the kidney’s filtration, regulation, and homeostatic functions fail — producing the fluid, electrolyte, acid-base, and toxin accumulation consequences that drive the clinical presentations and nursing priorities of acute and chronic kidney disease.
Acute Kidney Injury: Three Mechanisms of Filtration Failure
Acute kidney injury NCLEX pathophysiology classifies the cause of filtration failure by location: prerenal, intrinsic, or postrenal. Prerenal AKI results from inadequate renal perfusion — hypovolemia, heart failure, or sepsis reducing glomerular filtration pressure. The kidneys themselves are structurally intact; they simply lack sufficient blood flow to filter. Urine sodium is low and urine osmolality is high because the tubules avidly reabsorb sodium and water to restore circulating volume — the body’s normal response to hypoperfusion. Restoration of perfusion rapidly reverses prerenal AKI. Intrinsic AKI results from direct damage to kidney tissue — acute tubular necrosis from ischemia or nephrotoxins (aminoglycosides, contrast dye, NSAIDs, myoglobin from rhabdomyolysis) being the most common. Tubular cells are damaged and cannot perform their normal reabsorption functions. Urine sodium is high because damaged tubules cannot reabsorb it. Recovery requires days to weeks as tubular cells regenerate. Postrenal AKI results from urinary tract obstruction — benign prostatic hypertrophy, kidney stones, or tumor compressing the ureters. The back-pressure from obstruction reduces glomerular filtration. Relief of obstruction typically restores filtration rapidly. NCLEX pathophysiology nursing priorities across all AKI types include monitoring urine output as the primary filtration marker, managing hyperkalemia (the most immediately life-threatening electrolyte consequence of filtration failure), and identifying and removing the causative factor.
Chronic Kidney Disease: Progressive Loss and Adaptation
Chronic kidney disease NCLEX pathophysiology involves progressive loss of nephron function — the kidney’s structural and functional unit — through years of hypertension, diabetes, or inflammatory disease. As nephron mass decreases, the remaining nephrons undergo compensatory hyperfiltration — each remaining nephron filters at a higher rate than normal. This hyperfiltration sustains overall GFR temporarily but accelerates structural damage to the remaining nephrons, creating a progressive self-amplifying cycle of loss. When GFR falls below approximately 30 mL/min, clinical manifestations of retained solutes, fluid, and metabolic waste become pronounced: uremia (toxin accumulation causing encephalopathy, pericarditis, and platelet dysfunction), metabolic acidosis from inability to excrete hydrogen ions, hyperkalemia from reduced potassium excretion, hyperphosphatemia and hypocalcemia from disrupted vitamin D activation and phosphate excretion, and erythropoietin deficiency causing normochromic normocytic anemia. Every dietary restriction in CKD — potassium, phosphorus, sodium, and individualized protein restriction — is directly explained by these pathophysiological mechanisms.
The Renin-Angiotensin-Aldosterone System in Renal Disease
The RAAS is the central NCLEX pathophysiology mechanism connecting renal disease to hypertension, fluid retention, and cardiac damage. When renal perfusion decreases — from true volume depletion, heart failure, or intrinsic renal artery disease — juxtaglomerular cells release renin, which converts angiotensinogen to angiotensin I. Angiotensin-converting enzyme in the lungs converts angiotensin I to angiotensin II — a potent vasoconstrictor that raises blood pressure directly and stimulates aldosterone release from the adrenal cortex. Aldosterone causes sodium and water retention in the collecting duct, expanding volume. In renal failure, this system becomes chronically activated — contributing to the persistent hypertension of CKD that accelerates cardiovascular damage. This mechanism explains why ACE inhibitors and ARBs are the preferred antihypertensives in CKD: they directly interrupt RAAS activation, reducing both blood pressure and the proteinuric damage that angiotensin II causes to the glomerular filtration barrier.
Endocrine Pathophysiology: Hormones, Feedback, and Metabolic Crises
Endocrine NCLEX pathophysiology is organized around the feedback loop disruptions that produce the excess or deficiency states responsible for the most clinically dramatic — and most heavily tested — endocrine emergencies.
Diabetes: Insulin Deficiency and Its Metabolic Cascade
Diabetes NCLEX pathophysiology begins with insulin deficiency — absolute in type 1, relative in type 2 — producing a cascade of metabolic consequences. Without insulin, glucose cannot enter most cells, producing the paradox of cellular starvation in the face of serum hyperglycemia. Hyperglycemia exceeds the renal glucose reabsorption threshold, producing glycosuria — glucose in the urine — which draws water osmotically and produces polyuria. Volume depletion from polyuria stimulates thirst, producing polydipsia. Cellular starvation despite available glucose stimulates hunger, producing polyphagia despite hyperglycemia. In type 1 diabetes with absolute insulin deficiency, unopposed glucagon and stress hormones drive lipolysis — fat breakdown — producing free fatty acids that the liver converts to ketone bodies. Ketone accumulation produces metabolic acidosis — diabetic ketoacidosis — with the characteristic Kussmaul respirations (deep, rapid breathing compensating for metabolic acidosis by eliminating CO2) and fruity acetone breath from exhaled acetone. In type 2 diabetes, sufficient residual insulin prevents significant ketogenesis but inadequate insulin allows extreme hyperglycemia — producing the hyperosmolar state of HHS with profound cellular dehydration and neurological compromise without metabolic acidosis. Every nursing intervention for DKA and HHS is derivable from these mechanisms: isotonic saline restores intravascular volume, insulin drives glucose into cells, potassium must be replaced before or concurrently with insulin because insulin drives potassium intracellularly.
Thyroid Disorders: Metabolic Rate Disruption
Thyroid NCLEX pathophysiology affects every body system through the thyroid hormones’ role in regulating basal metabolic rate. Hyperthyroidism — excess thyroid hormone — accelerates all metabolic processes: heat production increases (hyperthermia, heat intolerance, diaphoresis), cardiac output increases (tachycardia, palpitations, atrial fibrillation in severe cases), weight loss despite increased appetite from accelerated catabolism, hyperreflexia from enhanced neuromuscular excitability, and anxiety and insomnia from central nervous system stimulation. Thyroid storm — the life-threatening extreme of hyperthyroidism — produces all of these findings at critical intensity, precipitated by physiological stress in a patient with uncontrolled hyperthyroidism. Hypothyroidism — thyroid hormone deficiency — produces the clinical opposite: hypothermia and cold intolerance, bradycardia, weight gain despite decreased appetite, hyporeflexia, constipation, and cognitive slowing. Myxedema coma — the life-threatening extreme of hypothyroidism — produces hypothermia, profound altered consciousness, and cardiovascular collapse. The NCLEX pathophysiology nursing recognition rule: all hyperthyroid signs are accelerations of normal function; all hypothyroid signs are decelerations. This one rule generates every clinical finding from the mechanism without separate memorization.
- Adrenal insufficiency NCLEX pathophysiology: Cortisol deficiency removes the permissive effect of cortisol on vascular smooth muscle tone and epinephrine synthesis, producing hypotension refractory to fluid resuscitation. Aldosterone deficiency causes sodium wasting and potassium retention — hyponatremia and hyperkalemia — with volume depletion and characteristic hyperpigmentation from elevated ACTH driving melanocyte stimulation. Addisonian crisis is treated with IV hydrocortisone, not simply fluid resuscitation.
- SIADH vs. diabetes insipidus: SIADH produces excess ADH, causing water retention without sodium retention — dilutional hyponatremia with inappropriately concentrated urine. Diabetes insipidus produces ADH deficiency or resistance, causing massive dilute urine output — up to 20 liters per day — with hypernatremia and extreme thirst. The pathophysiology is the exact inverse of SIADH, and the treatments are opposite: fluid restriction for SIADH, water replacement for diabetes insipidus.
Immune and Inflammatory Pathophysiology: Defense Gone Wrong
Immune and inflammatory NCLEX pathophysiology covers the mechanisms by which the body’s defense systems produce clinical harm when dysregulated — from the systemic inflammatory cascade of sepsis to the autoimmune destruction of specific tissues and the hypersensitivity responses of allergic disease.
Sepsis: The Dysregulated Inflammatory Response
Sepsis NCLEX pathophysiology begins with infection triggering an amplified systemic inflammatory response that extends far beyond the original site of infection. Pathogen recognition by immune cells activates cytokine release — particularly TNF-alpha, interleukin-1, and interleukin-6 — that produces widespread endothelial activation, increased vascular permeability, vasodilation, and coagulation system activation. Increased vascular permeability allows plasma to leak from the intravascular space into interstitial tissue — producing the clinical paradox of edema and hypovolemia simultaneously. Vasodilation drops systemic vascular resistance, causing the warm, flushed, bounding-pulse presentation of early distributive shock. Microvascular thrombosis from coagulation activation impairs oxygen delivery to tissues despite normal or high cardiac output — a state called distributive shock where blood flow is present but poorly distributed and oxygen cannot be extracted effectively. As sepsis progresses to septic shock, compensatory mechanisms fail, cardiac output falls, and multiorgan failure develops. The NCLEX pathophysiology one-hour bundle targets these mechanisms: cultures before antibiotics (avoid suppressing the pathogen before it can be identified), broad-spectrum antibiotics within one hour (reduce pathogen-driven inflammatory activation), IV fluid bolus (correct hypovolemia from vascular leak), and lactate measurement (quantify the severity of tissue hypoperfusion).
Anaphylaxis: IgE-Mediated Hypersensitivity
Anaphylaxis NCLEX pathophysiology is a type I IgE-mediated hypersensitivity reaction in which prior sensitization to an antigen produces IgE antibodies bound to mast cells and basophils. On re-exposure to the antigen, cross-linking of IgE antibodies triggers massive mast cell degranulation — the simultaneous release of histamine, leukotrienes, prostaglandins, and other mediators that produce the anaphylactic syndrome. Histamine causes vasodilation and increased vascular permeability — producing hypotension and edema — bronchospasm producing wheezing and respiratory distress, urticaria and angioedema from skin mast cell activation, and gastrointestinal cramping. The most dangerous manifestation is laryngeal edema — angioedema of the upper airway producing stridor and complete airway obstruction within minutes. The NCLEX pathophysiology emergency priority is epinephrine IM to the lateral thigh — not IV unless cardiac arrest is present — which reverses vasodilation through alpha-1 receptor activation, reduces bronchospasm through beta-2 receptor activation, and inhibits mast cell degranulation. Antihistamines and corticosteroids are important secondary treatments but not the initial priority because they act too slowly to reverse the immediate life threat.
Autoimmune Disease: Self-Directed Immune Attack
Autoimmune disease NCLEX pathophysiology involves the immune system failing to distinguish self from non-self, directing immune attack against host tissues. The mechanism varies by condition but the NCLEX pathophysiology principles are consistent: the immune attack targets specific tissues (joints in rheumatoid arthritis, myelin in multiple sclerosis, glomeruli in lupus nephritis), producing progressive tissue damage with episodes of acute exacerbation and partial remission. Corticosteroids suppress the inflammatory component of autoimmune attack — explaining their universal use in autoimmune flares despite significant side effects. Disease-modifying agents target specific components of the aberrant immune response. The NCLEX pathophysiology nursing priority in autoimmune disease is infection monitoring — the same immunosuppression that controls the autoimmune attack also blunts the response to genuine pathogens, making opportunistic infection the leading cause of morbidity in immunosuppressed patients.
Conclusion
NCLEX pathophysiology is not a separate content area to study in isolation — it is the mechanistic layer that makes every other clinical content area logically coherent rather than separately memorizable. When heart failure’s compensatory mechanisms explain every nursing intervention for that condition, when the Monroe-Kellie doctrine explains every positioning and monitoring priority for raised intracranial pressure, when insulin deficiency’s metabolic cascade explains every component of DKA management, and when the sepsis inflammatory cascade explains the one-hour bundle — clinical reasoning becomes derivable from mechanism rather than dependent on recall of disconnected facts under exam pressure.
Study NCLEX pathophysiology through mechanism-to-manifestation-to-intervention chains. For every condition, build the chain from the underlying physiological disruption through the compensatory mechanisms to the clinical signs they produce to the nursing interventions those signs call for. Practice generating these chains from memory using blank page recall after studying each system. When the mechanism is retrievable, the clinical knowledge that follows from it is derivable — and that derivability is what keeps clinical reasoning functional under the novel, complex scenario conditions the NCLEX is specifically designed to present.